ExaFEL
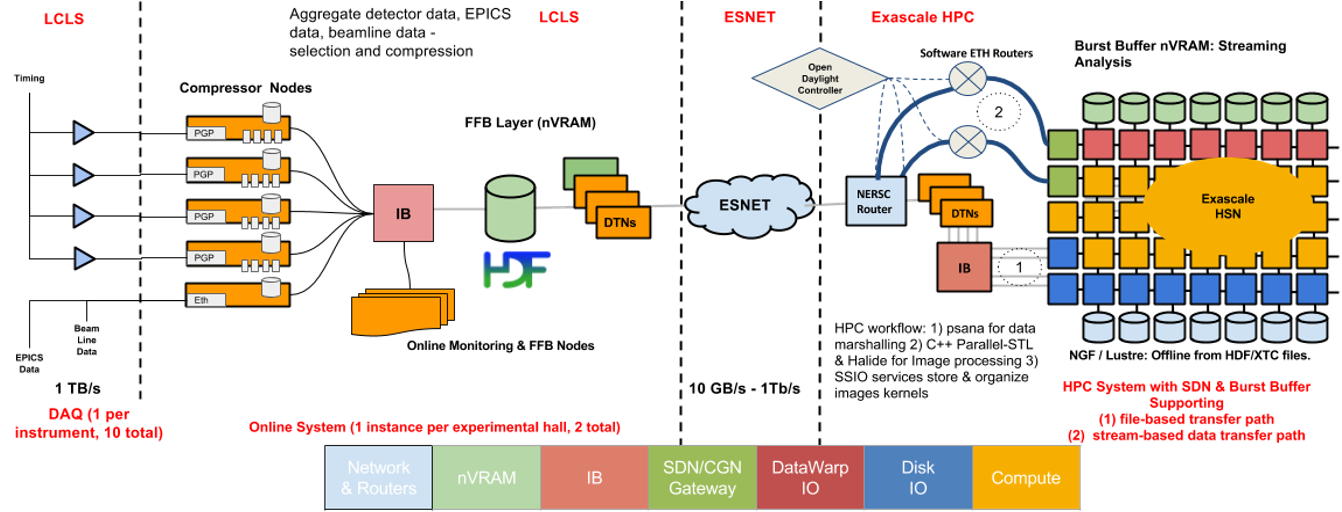
The ExaFEL project, part of the ECP’s exascale initiative, accelerates analysis of LCLS data, transforming weeks-long workflows into near-real-time (<10 min) results. By combining GPU-optimized algorithms (e.g., MTIP for single-particle imaging and nanoBragg for crystallography) with exascale workflows, it enables atomic-resolution reconstruction of dynamic molecular processes—such as enzymatic reactions or nanoparticle structural shifts—directly from burst-style diffraction data. The project integrates real-time data streaming from SLAC’s LCLS-II to supercomputers like NERSC and Summit, providing instant feedback to optimize experiments and resolve structural heterogeneities under physiological conditions, advancing fields from drug discovery to energy materials.
Key Objectives
- Develop an exascale-powered workflow for serial femtosecond crystallography (SFX) to rapidly merge billions of diffraction patterns into atomic-resolution 3D models.
- Create an automated single-particle imaging (SPI) pipeline using the Multi-Tiered Iterative Phasing (MTIP) algorithm to reconstruct dynamic molecular conformations from noisy 2D data.
- Enable real-time data streaming and feedback during experiments to optimize sample conditions and maximize beamtime efficiency.
- Scale computational workflows to match LCLS-II’s data rates, ensuring analysis keeps pace with ultrafast X-ray pulses.
Explore the ExaFEL project page
Partners & Collaborators
Amedeo Perazzo (PI, SLAC)
Johannes P Blaschke (LBNL/NERSC)
Robert Bolotovsky (LBNL)
Aaron S Brewster (LBNL)
Jeffrey Donatelli (LBNL/CAMERA)
Antoine DuJardin (SLAC)
Wu-chun Feng (Virginia Polytechnic Institute and State University)
Vidya Ganapati (LBNL)
Wilko Kroeger (SLAC)
Derek Mendez (LBNL)
Peter McCorquodale (LBNL)
Seema Mirchandaney (SLAC)
Christopher P. O’Grady (SLAC)
Daniel W. Paley (LBNL)
Frederic P Poitevin (SLAC)
Billy K Poon (LBNL)
Vinay B Ramakrishnaiah (LANL)
Nicholas K Sauter (LBNL)
Niteya Shah (Virginia Polytechnic Institute and State University, LANL)
Elliott Slaughter (SLAC)
Christine Sweeney (LANL)
Daniel Tchon ́(LBNL)
Monarin Uervirojnangkoorn (SLAC)
Felix Wittwer (LBNL/NSERC)
Michael E Wall (LBNL)
Chuck Yoon (SLAC)
Iris D Young (LBNL)
Publications
Blaschke, J. P., Bolotovsky, R., Brewster, A. S., Donatelli, J., DuJardin, A., Feng, W., et al. (2024). ExaFEL: extreme-scale real-time data processing for X-ray free electron laser science. Front. High Perform. Comput. 2. doi: 10.3389/fhpcp.2024.1414569
Blaschke, J. P., Brewster, A. S., Paley, D. W., Mendez, D., Bhowmick, A., Sauter, N. K., et al. (2024). “Real-time XFEL data analysis at SLAC and NERSC: A trial run of nascent exascale experimental data analysis,” in Concurrency and Computation: Practice and Experience.
Blaschke, J. P., Wittwer, F., Enders, B., and Bard, D. (2023). How a lightsource uses a supercomputer for live interactive analysis of large data sets. Synchrotron Radiat. News 36, 10–16. doi: 10.1080/08940886.2023.2245700
Chang, H.-Y., Slaughter, E., Mirchandaney, S., Donatelli, J., and Yoon, C. H. (2021). Scaling and acceleration of three-dimensional structure determination for single-particle imaging experiments with SpiniFEL. arXiv [Preprint]. arXiv 2109.05339.
Donatelli, J. J., Sethian, J. A., and Zwart, P. H. (2017). Reconstruction from limited single-particle diffraction data via simultaneous determination of state, orientation, intensity, and phase. Proc. Nat. Acad. Sci. 114, 7222–7227. doi: 10.1073/pnas.1708217114